Pharmacophore perception - PMapper
This manual gives you a walk-through on how to use the PMapper command line tool for pharmacophore fingerprint generation:
Introduction
PMapper perceives pharmacophoric propertiesof atoms of given molecular structures. When identified, pharmacophoric features are assigned (mapped) to corresponding atoms and stored along with the structure. This information can be used in searching large compound libraries for structures exhibiting similar therapeutic activity.
In this document a brief overview of pharmacophore feature identification is given first, while further documents describe how pharmacophores are used in various software tools developed by Chemaxon (like ScreenMD or JKlustor).
Usage
The general usage of PMapper as a command line tool is like:
pmapper [<options>] [<input files>]
Prepare the usage of the PMapper script or batch file as described in Preparing and Running Batch Files and Shell Scripts.
Alternatively, the PMapper class can be directly invoked as follows:
- Win32 / Java 2 (assuming that JChem is installed in c:\jchem):
java -cp "c:\jchem\lib\jchem.jar;%CLASSPATH%" \ chemaxon.pharmacophore.PMapper [<options>] [<input files>]- Unix / Java 2 (assuming that JChem is installed in /usr/local/jchem):
java -cp "/usr/local/jchem/lib/jchem.jar:$CLASSPATH" \ chemaxon.pharmacophore.PMapper [<options>] [<input files>]Options
The following options are available:
-h, --help this help message
-c, --config <filepath> path of the XML configuration file
-o, --output <filepath> output file path (default: stdout)
-t, --tag name of the SDFile tag to store the Pharmacophore Map (default: PMAP)
-S, --sdf-output SDF output (otherwise only PMAP list)
-g, --ignore-error continue with next molecule on error
-v, --verbose print calculation warnings to the console
The command line parameter --config is mandatory. This specifies the path and filename of a configuration file without which the program cannot operate. A detailed description of the format of this configuration file is given below.
The standardizer configuration can be given under a separate StandardizerConfiguration element node in the main configuration XML specified with the --config command line parameter. These transformations ensure an unambiguous molecule representation among the possible mezomer and tautomer forms of the same molecule. Other possible standardization include aromatization and hydrogenization. The standardization is only used to determine the pharmacophore points, but the original molecule will be written to the output. If this parameter is missing and no StandardizerConfiguration node is found in the configuration XML then no standardization is performed on the molecules. For a complete description of the standardization procedure and its configuration see the Standardizer Documentation.
If the command line parameter --ignore-error is specified, then import/export errors will not stop the processing but the error is written to the console and the molecule is skipped. By default, the program exits in case of molecule import/export erros.
Input
Most molecular file formats are accepted (e.g. MDL molfile, SDfile, SMILES).
If no input file name is given in the command line the standard input is read.
Output
If no output file name is given, results are written to the standard output.
Unless -P (or --PMAPonly ) is specified, the default output format is SDfile. The pharmacophore map tag is written into the output file, the default tag name is PMAP . This name can be changed by specifying the --tag option.
If the only output required is the pharmacophore map generated, then the --PMAPonly flag has to be specified.
Pharmacophore map data (value associated with the PMAP tag) can be visualized using MarvinView. In order to do so a palette definition property file has to be provided using the -p flag of mview and the name of the pharmacophore map flag (which is PMAP by default) has to be specified using the -t flag.
Configuration File
Pharmacophore maps generated by PMapper are substantially determined by the configuration file (specified following the --config mandatory command line parameter). The configuration file specifies
-
the pharmacophoric features to be perceived and mapped to atoms,
-
the rules that define these features,
-
further parameters that effect other pharmacophore related utilities (GenerateMD, ScreenMD).
The configuration file is an XML file.
The main pharmacophore point definition is given in the <Pharmacophores> section. Its <AtomSet> subsections define the pharmacophore point types: its unique id, its symbol used in the output as attributes and the definition expression in the text subsection.
The <Search> section specifies the substructure search attributes that are different from the default setting. See the Molecule Evaluator Documentation on Search Attributes for a precise description of the possible attributes and their default values.
The <Functions> section enables the user to add user-defined functions to the set of functions accessible from the expression strings: the <ID> attribute defines the reference name of the function, the <Class> specifies the function java class.
The <Mols> section defines molecule constants used in the pharmacophore definition expressions (e.g. as query structures). These molecule constants are either given by molecule files (strictly speaking paths and names of molecular files) or as smiles strings.
The <Plugins> section defines the calculation plugin modules used in quantitative approximation of chemical properties. The charge, pK a, log P calculations can be referenced in this way: the <ID> attribute defines the reference name of the plugin (this name is used in the expressions), the <Class> specifies the plugin java class and the subnodes provide the plugin parameters as name-value pairs.
Configuration examples
<!-- Pharmacophore configuration file -->
<PharmacophoreConfiguration Version ="0.3">
<Mols> <Mol ID="pos" Structure="../PD/Positive.mol"/>
<Mol ID="amine" Structure="../PD/Amine.mol"/>
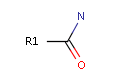
<Mol ID="amide" Structure="../PD/Amide.mol"/>
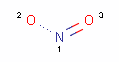
<Mol ID="nitro" Structure="../PD/Nitro.mol"/>
<Mol ID="hydrazide" Structure="../PD/Hydrazide.mol"/>
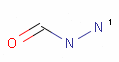
<Mol ID="amidine" Structure="../PD/Amidine.mol"/> </Mols>The molecular files referred in the above pharmacophore configuration XML file contain the molecular structures displayed below.
Here are some examples of molecular structures to be used as query substructures in the pharmacophore point identification procedure:
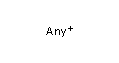 |
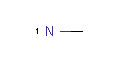 |
 |
|---|---|---|
 |
 |
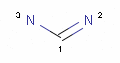 |
In these structural queries atoms may optionally be labeled with a map indices, which identify certain atoms of the pharmacophore group. Map indices must be unique among the atoms of a given molecule. Map indices can be used in pharmacophore type definition rules.
The <Pharmacophores> section is the pharmacophore definition section. In this section rules for every pharmacophore types can be defined. These rules combine the pharmacophore point feature definitions (query structures), preconfigured java method calls (molecule evaluator) including quantitative approximation of chemical properties by invoking calculator modules in logical expressions.
Definition examples
We provide two sample XML configurations.
-
pharma-frag.xml gives fragment-based definitions: the pharmacophore feature of an atom is determined by the functional groups containing the atom
-
pharma-calc.xml gives calculation-based definitions: the pharmacophore feature of an atom is determined by chemical feature calculations (e.g. partial charge, pKa) as well as containing functional groups
The calculation based configuration is shown in detail below.
<Search StereoCareChecking="false"/>In this example we set StereoCareChecking to false and leave all other attributes at their default values.
<Functions> <Function ID="data" class="chemaxon.util.expression.function.MolUtil"/> </Functions>The chemaxon.util.expression.function.MolUtil java class is configured to be referenced with the name data from the expression strings. For example, data("atomcount") returns the number of atoms in the input molecule. Here "atomcount" is a function argument of chemaxon.util.expression.function.MolUtil denoting the type of data enquiried.
<Pharmacophores> <AtomSet ID="Cationic" Symbol="+"> </AtomSet> </Pharmacophores>In the above example only one rule is introduced which combines several query structures to define the cationic pharmacophore feature. According to the rule an atom can be considered cationic if it matches the substructure defined in the Positive.mol or Amine.mol or Amidine.mol files, but not that in the Amide.mol and Nitro.mol files at the corresponding positions. More meaningfully, if it has a positive formal charge, or it is the nitrogen in an aliphatic amine or hydrazine, but it is neither an amide nor notro group nitrogen, or it is the carbon of either an amidine or guanidine group, but not carbamide carbon.
In the pharmacophore type definition rules map indices of the atoms can be referred after the ID (symbolic internal name) of the corresponding structure, separated by a semicolon, and in such case only the atom specified by the map index is subject to matching. Multiple atom map indices can also be used, these should be separated with comma characters. In the case when no map index is specified in the logical expression all atoms of the query are matched and evaluated.
A more complex example (pharma-calc.xml) is given below.
<Functions> <Function ID="formalcharge" class="chemaxon.util.expression.function.AtomProperties"> <Param Name="property" Value="charge"/> <Param Name="pH" Value="7"/> </Function> </Functions> <Plugins> <Plugin ID="acceptor" class="chemaxon.marvin.calculations.HBDAPlugin"> <Param Name="type" Value="acc"/> <Param Name="pH" Value="7"/> </Plugin> <Plugin ID="donor" class="chemaxon.marvin.calculations.HBDAPlugin"> <Param Name="type" Value="don"/> <Param Name="pH" Value="7"/> </Plugin> <Plugin ID="ioncharge" class="chemaxon.marvin.calculations.IonChargePlugin"> <Param Name="pH" Value="7"/> </Plugin> </Plugins>This is an example for calculator plugin decalaration with parameters. The IonCharge plugin calculates the distribution for the ionic forms of the input molecule, takes those forms for which the occurrence percentage is at least 1 percent at pH 7 taking into account maximum 8 ionizable atoms to avoid combinatorial explosure. The plugin sorts the atomic charge values on each ionic form according to descending occurrence percentage, the above declared min and max functions enable us to take the minimum or maximum charge value among these on each atom.
Take an example for a complete set of pharmacophore property definitions using this declaration:
<Mols> <Mol ID="arom" Structure="[*;a]"/> <Mol ID="cx" Structure="[C,F,Cl,Br,I,At]"/> </Mols> <Pharmacophores> <AtomSet ID="Aromatic" Symbol="r">arom</AtomSet> <AtomSet ID="Cationic" Symbol="+"> 0) || (ioncharge() > 0.4) ]]></AtomSet> <AtomSet ID="Anionic" Symbol="-"></AtomSet> <AtomSet ID="HydrogenBondDonor" Symbol="d"></AtomSet> <AtomSet ID="HydrogenBondAcceptor" Symbol="a"></AtomSet> <AtomSet ID="Hydrophobic" Symbol="h"></AtomSet> </Pharmacophores>This configuration defines the following pharmacophore properties:
-
Aromatic: an aromatic atom defined by a simple query.
-
Cationic: an atom with positive formal charge and partial charge greater than
0.4, both taken on the major microspecies at pH7, while -
Anionic: an atom with negative formal charge and partial charge less than
-0.4, both taken on the major microspecies at pH7. -
HydrogenBondDonor: determined directly by the HBDA plugin, taking the major microspecies at pH
7. -
HydrogenBondAcceptor: determined directly by the HBDA plugin, taking the major microspecies at pH
7. -
Hydrophobic: an aliphatic C, F, Cl, Br,I or At atom that is non of the above.
Note that in this case we reference previously defined atom sets with the atom set ID enclosed in {...} brackets (e.g. {Cationic} ). The reason for this is that atom sets are java.util.BitSet objects and without this special notation writing the ID of an atom set in an expression refers to the java.util.BitSet object itself. In the pharmacophore definitions above by {Cationic} we refer to the elements of the BitSet object, in other words we refer to those atom indices i for which Cationic.get(i) returns true. The {Cationic} notation is added to the standard expression syntax to enable the processing of this additional set-theoretic functionality.
Examples
-
A UNIX command that reads molecular structures from the standard input and writes result pharmacophore maps to the standard output:
cd test/pharmacophore pmapper -c Pharmacophores.xmlOne map per line is written on the output:
h;h;h;a h;h;h;a/d r;r;r;r;r;r;h;h;h d;h;h;+/d;-;-/a;-/a
-
A UNIX command that reads molecules given as SMILES strings from file nci10000.smiles located in the ./test/pharmacophore directory and writes results in the file named nci10000.sdf to be created in the same directory:
pmapper -S -c Pharmacophores.xml nci10000.smiles -o nci10000.sdf -
The same with a standardization procedure performed before pharmacophore mapping:
pmapper -S -c Pharmacophores.xml -s Standardizer.xml nci10000.smiles -o nci10000.sdf -
Processing an SDF file and displaying pharmacophore points using MarvinView:
pmapper -S -c Pharmacophores.xml med10000.sdf -o med10000-out.sdf mview -t PMAP -p Colors.ini med10000.sdfA screen-shot of MarvinView showing the structures with colored pharmacophore points:
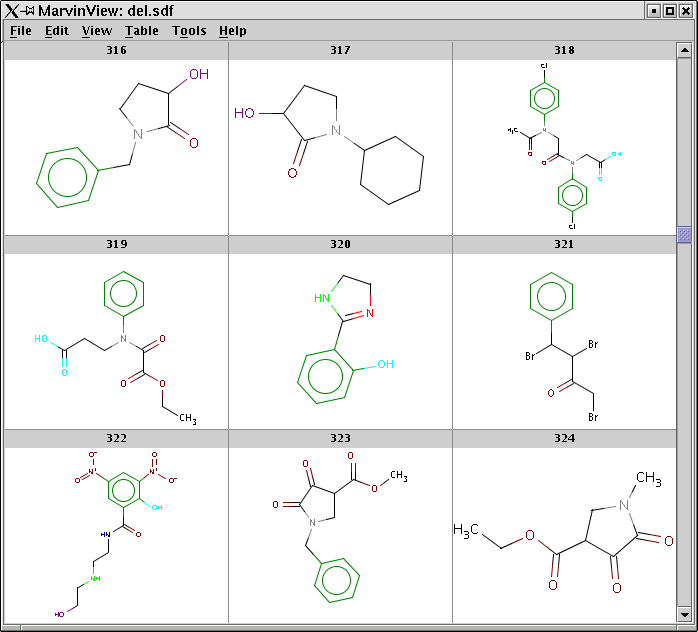
-
Same as above, but pharmacophore mapper results are piped directly into MarvinView:
pmapper -S -c Pharmacophores.xml med10000.sdf | mview -p colors.ini -t PMAP -{info}
-
Pharmacophore feature, pharmacophore property or pharmacophore type are alternative names.
-
Pharmacophore features are not necessarily directly assigned to atoms, for instance, the center of a benzene ring rather than its individual atoms can be characterised as aromatic. However, there is no evidence found in the relevant literature that such non-atom-based approaches have any advantages over atom-based descriptions.
-
For the sake of simplicity, only hydrogen bond donor and acceptor properties were used in the example. Apparently, there are no limitations to either the number or the kind of pharmacophore types used in the pharmacophore fingerprints.
-
Global effects that involve the entire molecule are also known, for instance tautomerism gives an obvious example of such mechanisms. These phenomena are not yet targeted in the present approach, however, intense work is under way to provide solutions to this particular problem.
-
References
-
Schneider, G.; Clement-Chomienne, O.; Hilfiger, L.; Schneider, P.;Kirsch, S.; Böhm, H-J. and Neihart, W. Virtual Screening for Bioactive Molecules by Evolutionary De Novo Design Angew. Chem. Int. Ed. 2000, 39, 4130-4133
-
Schneider, G.; Lee, M-L.; Stal, M. and Schneider, P. De novo design of molecular architectures by evolutionary assembly of drug-derived building blocks J. Comp-Aid. Mol. Des. 2000, 14, 487-494