Similarity search
Content
Introduction
Similarity searching finds molecules that are similar to the query structure. The similarity is calculated on the basis of the molecular descriptors or fingerprints of the chemical structures to compare. A molecular descriptor is a set of values associated with the molecular structure of the molecule. The term "molecular descriptor" refers to all kinds of structural keys, hashed fingerprints, binary fingerprints, different types of pharmacophore fingerprints and scalar values. There are various metrics available for the calculation.
JChem supports similarity search in files and in databases as well, and provides different hit display options for the visualization of hit sets.
Screen methods for similarity search
JChem Base product contains two types of built-in fingerprint methods: chemical hashed fingerprints for molecules and reaction fingerprints for reactions; and allows the use of more molecular descriptors/fingerprints as extended connectivity fingerprints , pharmacophore fingerprints , Burden eigenvalue descriptors ; and provides the possibility of the application of user-defined custom molecular descriptors.
Similarity search in file
Similarity search in files runs always on the basis of the built-in chemical hashed fingerprints for molecules and reaction fingerprints for reactions.
Similarity search in database
Similarity search in database could be run not only on the basis of chemical hashed fingerprints , but also on the basis of other screen methods as well. The possibility of the application of other screen methods strongly depends on the used platform.
Built-in fingerprint-based screen methods
JChem Base product contains the following built-in fingerprint-based screen methods available for similarity search:
The Chemical Hashed Fingerprints and Reaction Fingerprints are automatically generated in the JChem tables during the data import process; similarity search uses these generated fingerprints by default.
The built-in fingerprints are generated on-the-fly if similarity search is executed in files.
Descriptor-based screen methods
The following descriptor-based screen methods are available for similarity search:
-
Extended Connectivity Fingerprints ( ECFP/FCFP )
-
Burden eigenvalue ( BCUT ) descriptors
For the application of descriptor-based screen methods separate license is needed.
For the generation of the above listed fingerprints/descriptors, you have to add them to the tables concerned, and let the tables be recalculated. See Administration guide of JChem Manager about how to modify a JChem table by addition molecular descriptors. Molecular descriptors can be generated on the basis of molecular descriptor configuration files. See an example configuration file here .
Further information about generation of molecular descriptors in JChem Cartridge can be found in Generic Molecular Descriptor support in JChem Cartridge, and about the GenerateMD application in Generate Molecular Descriptors documentations.
Molecules in low energy conformation serve as input for 3D similarity comparison. 3D conformation may be obtained by ChemAxon's Clean3D tool.
Screen3D evaluates 3D Tanimoto similarity between pairs of molecules by maximizing the intersection of their volumes. The volume may colored by pharmacophoric properties or by atom types. During the calculation the structures are translated and rotated and the rotatable bonds are tweaked. See example here.
{kind=link}
Descriptor-based similarity search can be speeded up by caching the descriptor data. See details of caching.
Customized screen methods
In JChem there is a possibility to apply user-defined custom descriptors/fingerprints. See the details in the Custom descriptor implementation section of the JChem developer's guide, Generic Molecular Descriptor support in JChem Cartridge, and Generate Molecular Descriptors documentations.
Metrics
Similarity / Dissimilarity metrics for molecules
Various metrics are provided in JChem to compute the value of similarity or dissimilarity. Some metrics (for example Tanimoto) provide similarity values, some other metrics (for example Euclidean) provide dissimilarity values. The values calculated with the metrics listed in the table below (with the exception of Euclidean) vary from 0 to 1. Similarity (S) value can be calculated from the value of dissimilarity(D): S = 1 - D (with the exception of Euclidean metric).
The larger the value of the dissimilarity coefficient the bigger the difference between the two structures is.
Notes: different molecules may have 0 dissimilarity, if their descriptors are the same.
The table below lists the dissimilarity metrics and their general formulas; the representation of the formulas is based on finite length binary fingerprints.
Notations :
| NA | number of bits set in the fingerprint of molecule A |
|---|---|
| NB | number of bits set in the fingerprint of molecule B |
| NA&B | number of bits set in the fingerprint of both molecules A and B |
| α | coefficient representing the weight of properties of molecule A, its value is between 0 and 1 |
| β | coefficient representing the weight of properties of molecule B, its value is between 0 and 1 |
| Metric | Dissimilarity formula |
|---|---|
| Tanimoto | 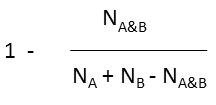 |
| Tversky | 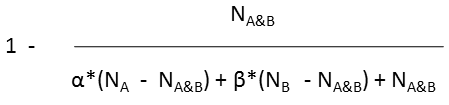 |
| Substructure (extreme case of Tversky: α=0, β=1) | 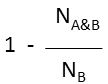 molecule B as substructure of molecule A molecule B as substructure of molecule A |
| Superstructure (extreme case of Tversky: α=1, β=0) | 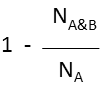 molecule B as superstructure of molecule A molecule B as superstructure of molecule A |
| Euclidean | 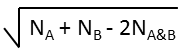 |
Reaction fingerprint metrics
Two types of reaction similarity calculations have been introduced: structural and transformational. Structural distinguishes the reactant and the product sides, while transformational relates to three levels of coarseness. With these considerations five metrics need to be introduced to efficiently estimate the five different categories of reaction similarity. These metrics are as follows:
-
ReactantTanimoto
-
ProductTanimoto
-
StrictReactionTanimoto
-
MediumReactionTanimoto
-
CoarseReactionTanimoto
See details of Reaction fingerprint metrics .
Hit display
The visualization of the search results is a very important feature of the JChem products. Hit display options in similarity search involve coloring of maximum common substructures and the selection of what to display (similarity or dissimilarity score, query structure, other labels and boxes). See the description of hit display options available in similarity search here.
How to perform similarity search
Similarity search is performed on the basis of the built-in molecular descriptors ( chemical hashed fingerprints for molecules and reaction fingerprints for reactions) by default. As described above, in case you want similarity search to be performed on the basis of other molecular descriptors/fingerprints you have to add these descriptors to the target tables concerned before the search.
For the execution of similarity search you have to specify:
-
search type as similarity search;
See how to set search type on different platforms (Instant JChem, JChemBase API, JChem Cartridge, jcsearch command line).
-
the desired molecular descriptor/fingerprint - if they were added to the target table or to the target file;
-
the desired dissimilarity metric;
See how to set dissimilarity metric on different platforms (Instant JChem, JChemBase API, JChem Cartridge, jcsearch command line).
-
the desired (dis)similarity threshold;
See how to set (dis)similarity threshold on different platforms (Instant JChem, JChemBase API, JChem Cartridge, jcsearch command line).
-
the desired hit display options.
Hit display/coloring possibilities of hits (displaying query structure, displaying similarity or dissimilarity score, displaying other labels and boxes) in similarity search are presented here .
See how to set hitdisplay options for similarity search (similarity, query display, display labels and boxes) on different platforms (Instant JChem, JChemBase API, JChem Cartridge, jcsearch command line) and how to set coloring options (coloring, hit color, non-hit color) on different platforms.
Examples
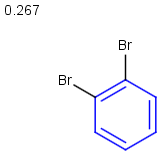
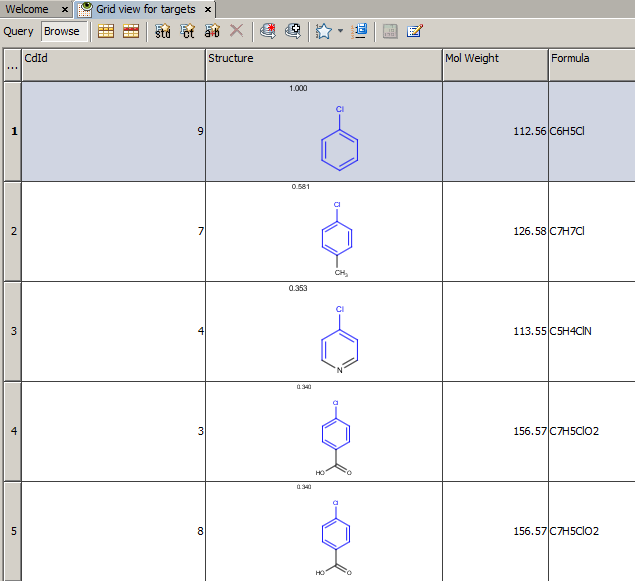
- Example of running similarity search in Instant JChem with default Chemical Hashed Fingerprint, Tanimoto metric, and Similarity threshold set to 0.3:
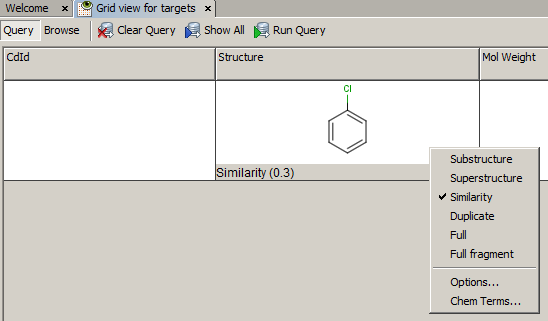
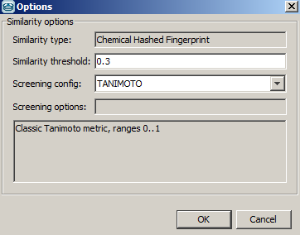
Result:
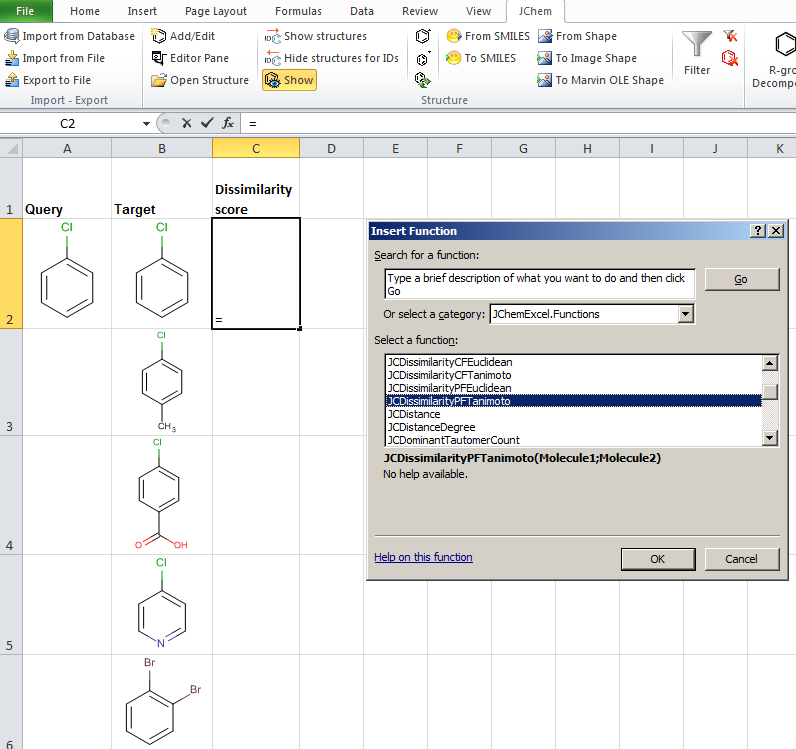
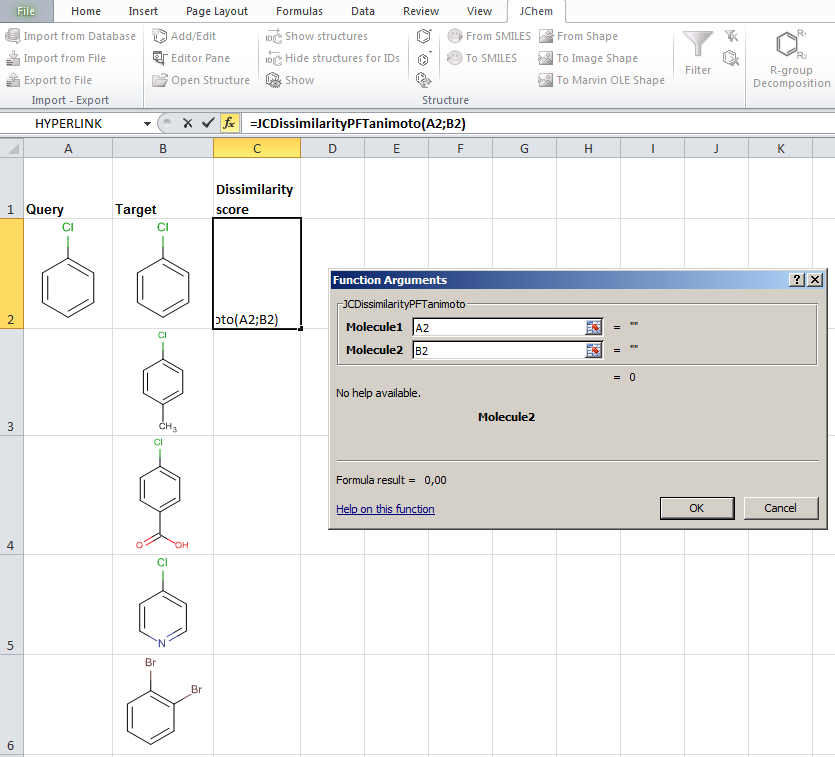
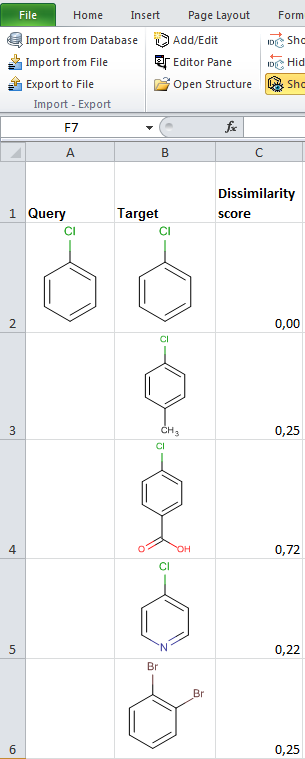
- JChem for Excel example for determining dissimilarities calculated with Tanimoto distances based on a pharmacophore fingerprint. The custom JChem Excel Function, namely JCDissimilarityPFTanimoto is applied.
-
See examples for similarity search in JChem Cartridge here.
-
Example of running similarity search in command line with jcsearch
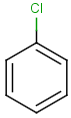
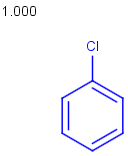
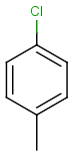
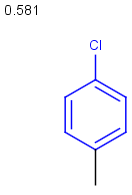
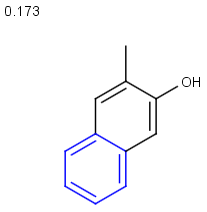
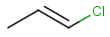
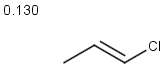
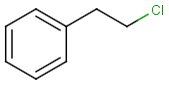
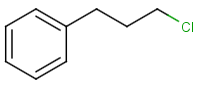
Table below illustrates similarity search (performed with default search options with the built-in Tanimoto metric and dissimilarity threshold set to 0.9). Similarity score is shown, and the maximum common substructures are highlighted in the hit structures. Similarity search was performed in file with jcsearch :
jcsearch -q query.mrv targets.mrv -t:i:0.9 --hitColoring:y -f mrv -o hits.mrvQuery Targets Similarity score based on CFP/ Hits