logP Plugin¶
This manual gives you a walk-through on how to use the logP Plugin.
Table of Contents¶
Introduction¶
The logP Plugin calculates the logarithm of the octanol/water partition coefficient (logP), which is used in QSAR analysis and rational drug design as a measure of molecular lipophylicity/hydrophobicity.
Calculation method¶
The calculation method is based on the publication of Viswanadhan et al. The logP of a molecule is calculated based on the atomic logP contributions. However, the algorithm described in the mentioned paper was extended with the followings:
-
Many atom types were re-defined to accommodate electron delocalization. Contributions of ionised atoms/fragments were added.
-
The logP of zwitterions are calculated as their logD value at the isoelectric point.
-
The effect of hydrogen bonds on the logP is considered if there is a chance to form a six membered ring between suitable donor and acceptor atoms. The effect is added to the sum of atomic logP contributions as a structural increment.
-
New atom types were introduced, especially for S, C, N and metal atoms.
To read more about the formal definition, derivation and the calculation of logP and logD, see the following page.
Calculation result¶
The result of the logP calculation appears in a new window, either in a MarvinView (for 2D display) window or in a MarvinSpace (for 3D display) window.
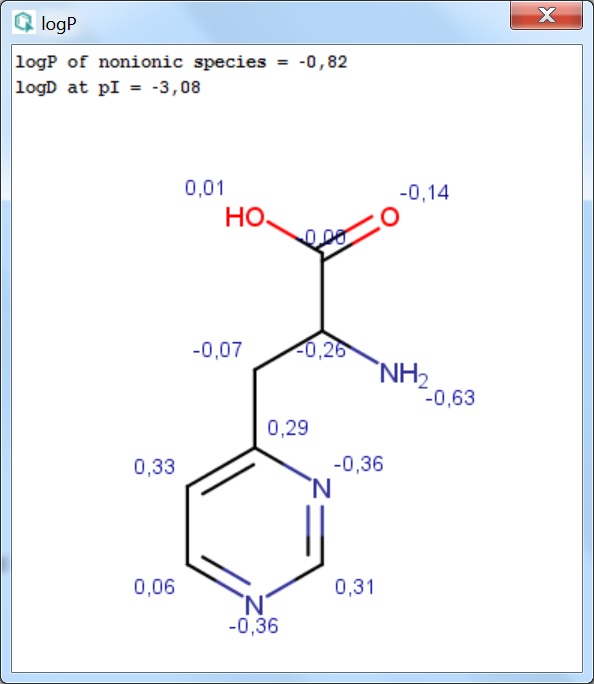
Fig. 1 The logP result window with atomic increments displayed in MarvinView
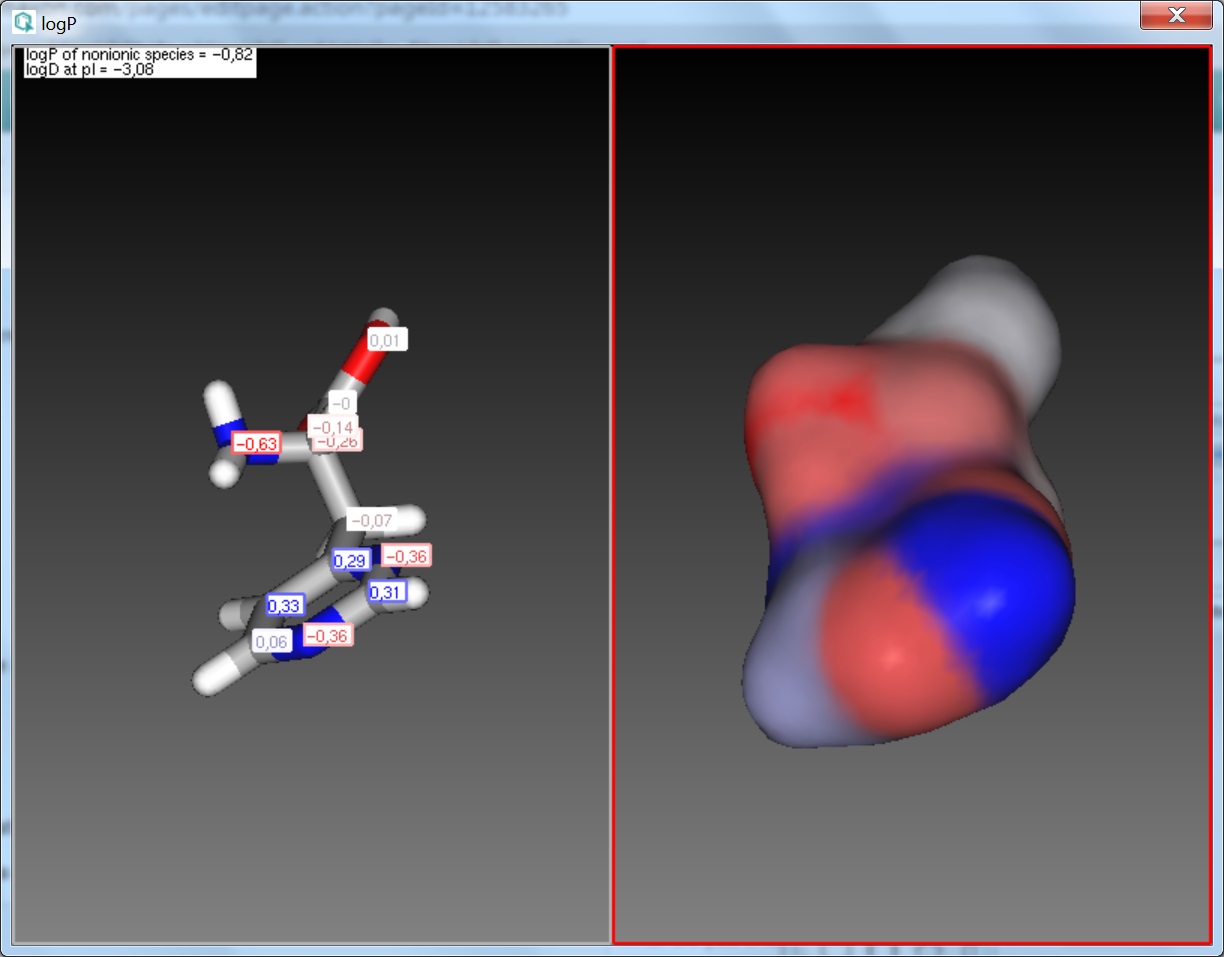
Fig. 2 The logP result window with atomic increments displayed in MarvinSpace
Displayed values¶
The calculated and displayed logP value(s) depend(s) on the input structure:
- logP of ionic species: this logP value is returned if the input structure is charged.
- logP of nonionic species: this is the logP value of the neutral form of the input structure. If a neutral molecule is
used as input, the non-ionic logP is returned.
- logD at pI: in case of zwitterionic input structures the logD at the isoelectric point (pI) is returned.
- Structural increment: the additional logP increment coming from the internal structure of the input molecule.
The position of atoms relative to one another can have secondary effects on the distribution of the molecule between the octanol and water phases. These are typically intra-molecular effects, e.g. H-bonds, effects of ionized groups onto each other etc. The sum of these effects is expressed as a structural increment, which is added to the sum of atomic logP contributions. This increment is usually fine-tuned in our model to best describe the secondary effects of intra-molecular forces.
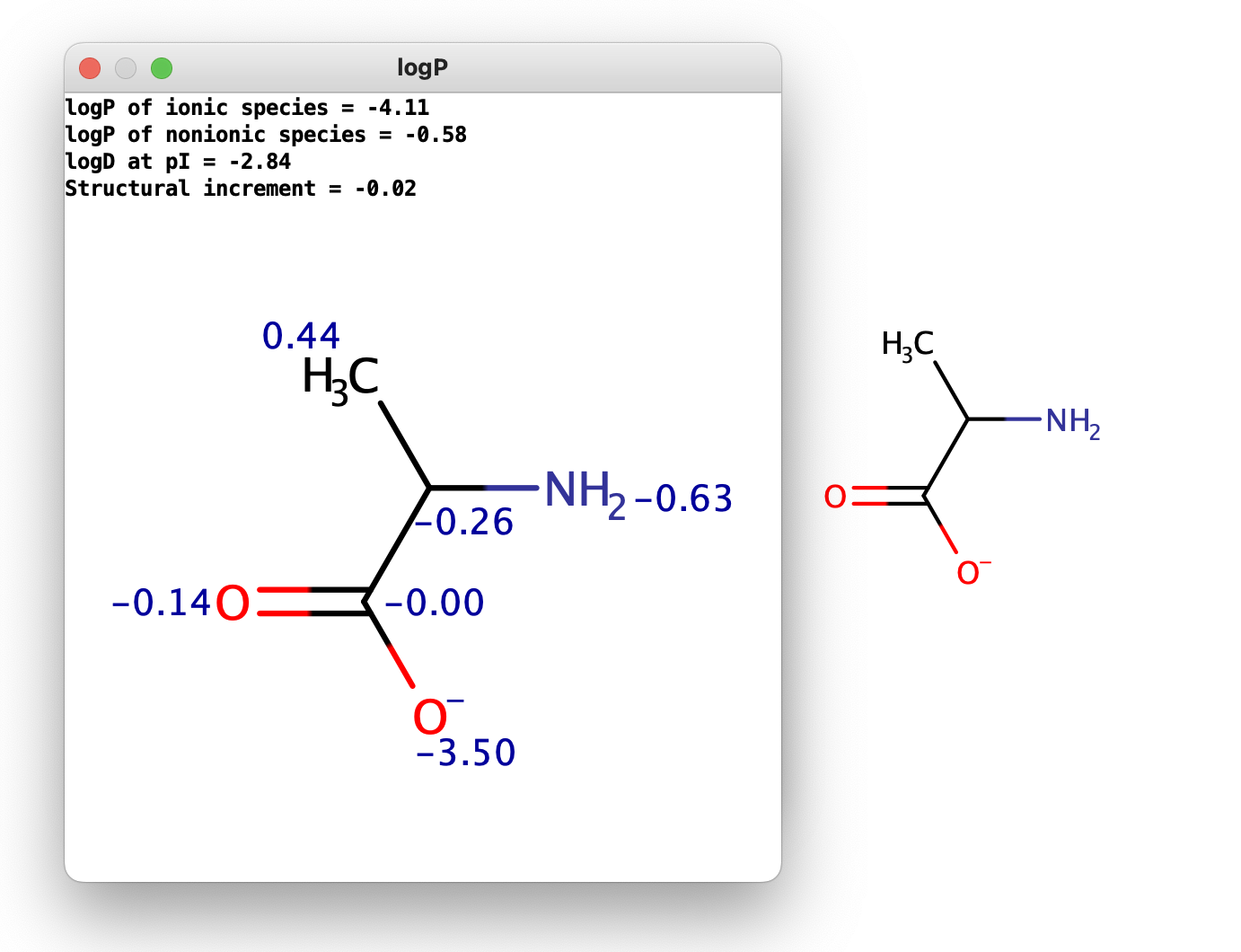
Fig. 3 The displayed logP results for a charged input structure
Options¶
The following options can be set in the logP Plugin.
General Options¶
These options are related to the calculation of logP.
Method¶
This option defines the logP calculation method. The following methods are available:
- Consensus: this method is based on a consensus model built from the Chemaxon model, the Klopman et al. model and the PhysProp database.
- Chemaxon: this method is based on Chemaxon's own logP model, which is an extension of the VG method (derived from Viswanadhan et al.).
You can read more about it here.
- User defined: if a molecule set and corresponding experimental logP values are available, it can be used for training the available logP methods. See this page on creating a training library.
The Consensus logP method is a unique, in-house developed logP model based on the methods listed above. It is method is similar (but not identical) to the ClogP method, while the Chemaxon logP method is similar (but not identical) to the AlogP method.
Training ID¶
This option lists all logP training libraries generated by cxtrain. The option becomes active only if the User defined calculation method is selected.
Electrolyte concentration¶
These are options for setting the anionic and cationic concentrations for the calculation.
- Cl- concentration: can be set between 0.1 and 0.25 mol/L.
- Na+ K+ concentration: can be set between 0.1 and 0.25 mol/L.
Consider tautomerization/resonance¶
The logP is calculated for the major tautomer of the input structure if this option is enabled.
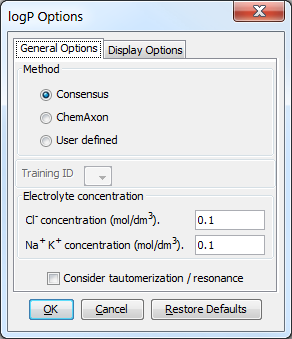
Fig. 4 The logP Options window showing the General Options panel
Display Options¶
These options are related to the display of the logP calculation result.
Precision¶
The precision of the calculated logP can be set by the number of decimal places to be displayed. The default is 2.
Show value¶
It sets the type of logP result(s) to be displayed. These can be:
- Increments: displays the atomic and structural increments.
- logP: displays the logP value(s).
Display in MarvinSpace¶
This option displays the results in a 3D MarvinSpace window. If disabled, the results will show in a 2D MarvinView window.
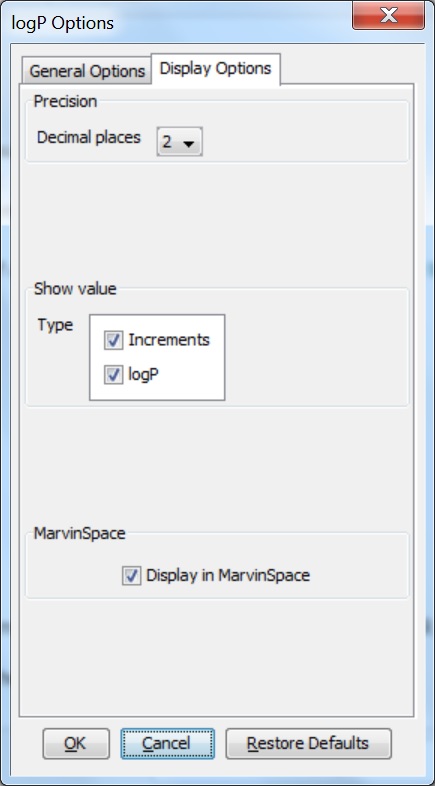
Fig. 5 logP Options window with the Display Options panel
References¶
-
Viswanadhan, V. N.; Ghose, A. K.; Revankar, G. R.; Robins, R. K., J. Chem. Inf. Comput. Sci., 1989, 29, 163-172; doi
-
Klopman, G.; Li, Ju-Yun.; Wang, S.; Dimayuga, M., J.Chem.Inf.Comput.Sci., 1994, 34,752; doi
-
PHYSPROP© database
-
Csizmadia, F; Tsantili-Kakoulidou, A.; Pander, I.; Darvas, F., J. Pharm. Sci., 1997, 86, 865-871; doi