Structure Display Options in JChem for Excel
With these options, it is possible to define the style and chemistry-related properties of the displayed structures.
For more information, see the Structure Display Options section of the MarvinSketch User's Guide.
The figure below shows the structure display options of JChem for Excel.
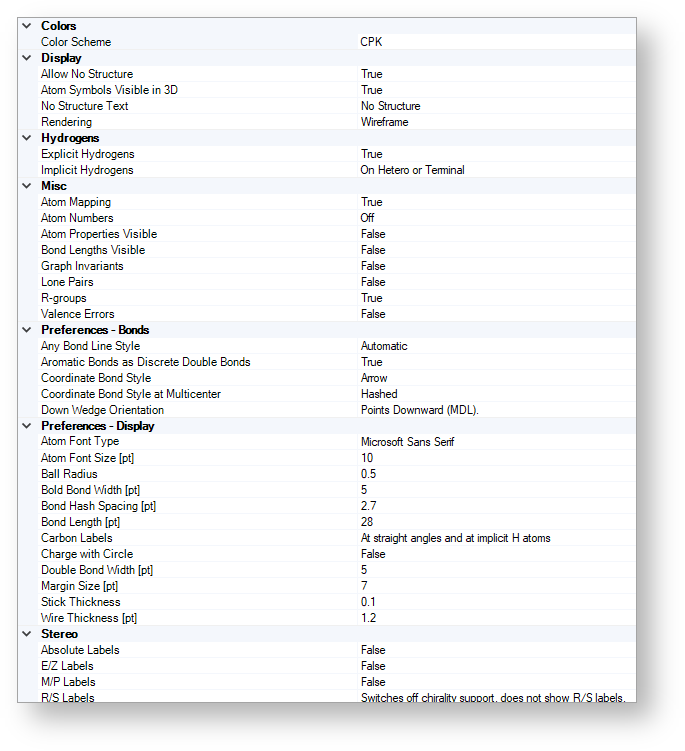
Colors
Color Scheme
Use this option to specify the color scheme of the displayed structures.
The following schemes are available:
-
Monochrome -
CPK -
Shapely -
Group
Display
Allow No Structure
This option affects cell behavior when you click Add/Edit and leave the canvas of the structure editor empty then click OK.
By using this option, you can specify whether the No Structure text is added to cells after the scenario described above or not. When the text is added to the cell, the JCSYSStructure function is also created for the cell. You can specify the text string by using the No Structure Text option.
The available values for this option are as follows:
-
TRUE -
FALSE
Atom Symbols Visible in 3D
Use this option to specify whether atom symbols are visible in 3D mode or not.
{info} Atoms may become invisible in wireframe mode when atom symbols are hidden.
The available values for this option are as follows:
-
TRUE -
FALSE
No Structure Text
Use this option to customize the text that is displayed when no structure is drawn to a cell.
The default text is No Structure.
Rendering
By using this option you can specify the structure display style.
The following styles are available:
-
Wireframe -
Ball and Stick -
Spacefill
The default style is Wireframe.
The table below shows the different styles.
| Style | Wireframe | Ball and Stick | Spacefill |
|---|---|---|---|
| Example | 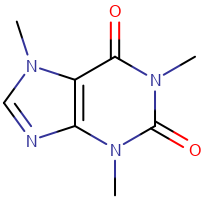 Caffeine Wireframe Caffeine Wireframe |
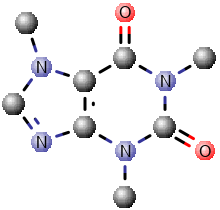 Caffeine Ball and Stick Caffeine Ball and Stick |
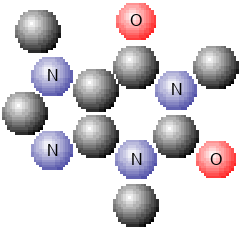 Caffeine Spacefill Caffeine Spacefill |
Hydrogens
Explicit Hydrogens
Use this option to specify the visibility of explicit hydrogen atoms.
The available values for this option are as follows:
-
TRUE -
FALSE
Implicit Hydrogens
Use this option to specify the display mode of implicit hydrogens.
The following display modes are available:
-
Off -
On Heteroatoms -
On Heteroatoms or Terminal -
All
The default display mode is On Heteroatoms or Terminal.
The table below shows the different display modes.
| Display mode | Off | On Heteroatoms | On Heteroatoms or Terminal | All |
|---|---|---|---|---|
| Example | 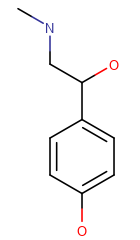 |
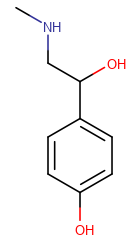 |
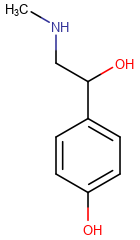 |
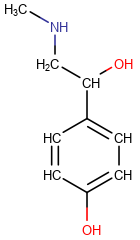 |
Misc
Atom Mapping
Use this option to specify whether atom mapping is shown or not.
The available values for this option are as follows:
-
TRUE -
FALSE
Atom Numbers
By using this option you can select from the following atom number displays:
-
Off:No atom numbers are displayed on the structures. -
AtomNumbers:Atom numbers are ordered as in Marvin. -
IUPACNumbering:Atom numbers are ordered according to IUPAC rules.
The default display is Off.
The table below shows the various display options.
| Display mode | AtomNumbers | IUPACNumbering |
|---|---|---|
| Example | 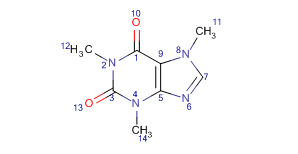 |
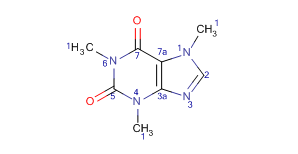 |
Atom Properties Visible
Use this option to specify whether atom properties are displayed or not.
The available values for this option are as follows:
-
TRUE -
FALSE
Bond Lengths Visible
Use this option to specify whether bond lengths are visible or not.
The available values for this option are as follows:
-
TRUE -
FALSE
Graph Invariants
Use this option to specify whether graph invariants are displayed or not.
The available values for this option are as follows:
-
TRUE -
FALSE
Lone Pairs
Use this option to specify whether lone pairs are visible or not.
The available values for this option are as follows:
-
TRUE -
FALSE
R-groups
Use this option to specify whether R-group definitions are visible beside Markush or scaffold structures containing R-groups or not.
The available values for this option are as follows:
-
TRUE -
FALSE
Valence Errors
Use this option to specify whether valance errors are shown or not.
The available values for this option are as follows:
-
TRUE -
FALSE
Preferences – Bonds
Any Bond Line Style
By using this option, you can specify the display mode of bonds with unknown order.
The following modes are available:
-
Automatic(in most cases, this means a dashed line, solid lines are only used when all bonds are generated from atom coordinates, for example, in the case of XYZ and PDB files). -
Dashed
Aromatic Bonds as Discrete Double Bonds
Use this option to specify whether aromatic structures are displayed in Kekule format (discrete double bonds instead of the aromatic rings) or not.
The available values for this option are as follows:
-
TRUE -
FALSE
Coordinate Bond Style
By using this option, you can specify the line style of coordinative bonds when both atoms are single.
The following styles are available:
-
Arrow -
Solid
Coordinate Bond Style at Multicenter
By using this option, you can specify the line style of coordinative bonds when one of the atoms is multicenter.
The following styles are available:
-
Hashed -
Solid
Down Wedge Orientation
Use this option to set the display convention of the wedge bonds.
The following displays are available:
-
Points Downward (MDL) -
Points Upward (Daylight)
The default display is Points Downward (MDL).
Preferences – Display
In the case of these options, when defining a numerical value use positive values (in some cases zero is allowed). A negative value is overwritten with the default value.
Atom Font Type
Use this option to specify the font type of atoms in the structures.
The available types depend on the regional settings of your operating system. It is possible to use non-default Windows font types.
The default style is Microsoft Sans Serif.
Atom Font Size [pt]
Use this option to specify the atom label font size in points.
Ball Radius
Use this option to specify the ball radius for the ball and stick model.
Bold Bond Width [pt]
Use this option to specify the width of the line (in points) used when a bold bond is drawn.
Bond Hash Spacing [pt]
Use this option to specify the spacing between (in points) the hashed lines for single down wedged bonds and hashed bonds.
Bond Length [pt]
Use this option to specify the length of bonds in points.
Any positive number can be used.
Carbon Labels
By using this option, you can specify how carbon labels are displayed.
The following modes are available:
-
Always -
Never -
At straight angles and implicit H atoms
The default option is A t straight angles and implicit H atoms.
The table below shows the different modes.
| Display mode | Always | Never | At straight angles and implicit H atoms |
|---|---|---|---|
| Example | 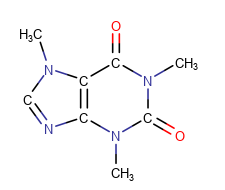 |
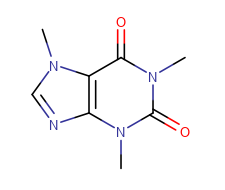 |
 |
Charge with Circle
Use this option to specify whether charges on molecules are displayed in a circle or not.
The available values for this option are as follows:
-
TRUE -
FALSE
Double Bond Width (pt)
Use this option to specify the width of double bonds in points.
Margin Size (pt)
Use this option to specify the margin width of the bounding rectangle in points.
Stick Thickness
Use this option to specify the stick diameter for the ball and stick model.
Wire Thickness (pt)
Use this option to specify the bond thickness in points for the wireframe model.
Stereo
Absolute Labels
Use this option to specify whether the absolute stereo configuration labels are visible or not.
The available values for this option are as follows:
-
TRUE -
FALSE
E/Z Labels
Use this option to specify whether the absolute double bond stereo configuration labels are visible or not.
The available values for this option are as follows:
-
TRUE -
FALSE
M/P Labels
Use this option to specify whether the axial stereochemistry labels are visible or not.
The available values for this option are as follows:
-
TRUE -
FALSE
R/S Labels
The table below lists all possible values for this option along with a description and the name of the corresponding MarvinSketch option.
| Option in JChem for Excel | Description | Option in MarvinSketch |
|---|---|---|
| Switch off chirality support, do not show R/S labels | No R/S labels are displayed. | Stereo > R/S Labels > None |
| Show R/S if the chiral flag is set for the molecule | R/S labels are displayed if an absolute stereo (chiral) flag is added to the molecule. | Stereo > R/S Labels > Absolute Stereo |
| Show R/S for any molecule | Shows R/S chirality for all atoms in the molecule. | Stereo > R/S Labels > All |
| Show all possible R/S for any molecule | Atoms that could have an R/S stereo label—but it is not possible to decide which one exactly—are marked with a question mark. | Stereo > R/S Labels > All possible |
For more information about these options in MarvinSketch, see View Menu.