Comparison of JChem PostgreSQL Cartridge and JChem Oracle Cartridge
This document compares the main functional differences between JChem Oracle Cartridge (JOC) and JChem Postgres Cartridge (JPC) and their reasons.
Introduction
The main concept of JPC is to provide a cost effective and improved alternative to JOC.
Knowing the weaknesses of JOC, like complicated option space, embarrassing authentication we were determined to find a better solution.
Architecture
JOC : Because of the architecture of the JChem Oracle Cartridge system, it is necessary to use the jchem_core_pkg.use_password() function in order to be able to execute operations on JChem server’s side.
JPC : JChem Postgres Cartridge architecture does not require this kind of identification.
Tables, indexing
JOC : handles regular Oracle tables and JChem tables. The CREATE INDEX statement using indextype jc_idxtype generates index tables, which make JChem be able to function (e.g., be able to run searches).
JPC : does not handle JChem tables; exclusively regular Postgres tables are handled. The column for storing the chemical structures must be Molecule type. CREATE INDEX using chemindex or sortedchemindex makes the JChem search processes run faster; however, searching in unindexed tables is also possible.
Import
JOC : Import function only into JChem tables is provided, but not into regular Oracle tables.
JPC: SDF file import into regular Postgres tables is supported. See detailed information in JChem PostgreSQL Cartridge Manual.
Table settings / CREATE INDEX parameters in JOC vs. molecule types in JPC
JOC : The table settings of JChem tables define how the molecules in the table will be interpreted:
-
table types molecule, any structure, reaction, query structure, markush library
-
standardization (default or customized)
-
assume absolute stereo (default: yes, but can be set to ‘no’)
-
filter out duplicates
-
duplicate search uses tautomers
In the case of regular Oracle tables the above settings can be applied as CREATE INDEX parameters.
JPC : There are no table settings.
-
table types do not exist
-
The required standardization has to be set in the molecule type files. The name of the molecule type file has to be applied as a parameter of the Molecule column type. See details in JChem PostgreSQL Cartridge Manual.
-
assume absolute stereo - from version 20.12 stereoAssumption=RELATIVE is also supported. Former versions assume only absolute stereo
Search engine
JOC : search engine of JChem Base is used
JPC : search is based on a newly developed search engine
Search types
JOC : search types can be set using the t parameter of the jc_compare operator; duplicate, substructure, full structure, full fragment, superstructure, and similarity search are supported
JPC : duplicate, substructure, full fragment, superstructure, and similarity search are supported. Full structure search is not supported.
JPC provides the following operators (see details in JChem PostgreSQL Cartridge Manual)
| Search type | Operator | Comment | ||||||
|---|---|---|---|---|---|---|---|---|
| duplicate | = | |||||||
| substructure | < | |||||||
| full fragment | < | same as substructure, but query must be transformed as query_transform(query_structure, 'fullfragment') | ||||||
| full fragment | <= | from version 20.15 | ||||||
| superstructure | > | see details | ||||||
| similarity | ~ | , | <~ | , | ˇ> |
Markush structures
JOC : Markush search (searching Markush targets) and using query structures with Markush features are supported.
JPC : Markush search (searching Markush targets) is not supported. Using query structures with Markush features - with the exception of homolgy groups - is supported.
Similarity search
JOC : Similarity search based on Chemical hashed fingerprints and Tanimoto metric works by default. It is possible to use some built-in descriptors and to use custom descriptors. A few additional metrics are provided as well.
JPC : only Chemical hashed fingerprints with Tanimoto metric is supported (at the moment).
The performance of the similarity search is much better compared to JOC.
See detailed steps described in JChem PostgreSQL Cartridge Manual.
The similarity value between two structures can be different in JOC and in JPC because their default fingerprint settings are different as shown in the table below:
| Fingerprint property | JPC | JOC |
|---|---|---|
| Fingerprint length | 512 | 512 |
| Bits to be set for patterns | 1 | 2 |
| Maximum pattern length | 6 | 6 |
Tautomer handling
Basic differences:
| Feature | JPC | JOC | Comment |
|---|---|---|---|
| default setting | type of the column storing the structures defines whether tautomer search is switched on or not;available molecule types are found under /etc/chemaxon/types/;you can add, modify, and delete molecule type files according to your needs;tautomer mode set in a molecule type file can be OFF (tautomers are not taken into account) or GENERIC (tautomer search runs on the basis of generic tautomers) | tautomer search OFF-- with the exception of indexes created with 'TDF:y' parameter where tautomer search ON | |
| tautomer substructure search | the query is compared to the generic tautomer of the target | all tautomer enumerants of the query are compared to the target | see hit difference examples in the next table |
| tautomer full fragment search | the query is compared to the generic tautomer of the target | the generic tautomer of the query is compared to the generic tautomer of the target | no hit differences are expected between JPC and JOC |
Hit differences are expected in tautomer substructure search between JOC and JPC.
Examples:
| Query | Target | JOC hit | JPC hit |
|---|---|---|---|
| No | Yes | ||
| Yes | No | ||
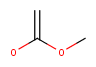 |
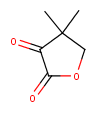 |
Yes | No |
The CANONIC_GENERIC_HYBRID mode of tautomer search in JPC works in fullfragment and in duplicate search similarly as tauromer search with tautomerEqualityMode=nc in JOC.
Search options
JOC : search options can be used as parameters of the jc_compare operator
JPC : At the moment only the following search options can be applied:
-
modifying double bond stereo interpretation
-
ignore tetrahedral stereo information (available from version 5.1)
-
tautomer search can be executed in structure tables where the molecule type of the structure column has tautomer = GENERIC setting
Our aim is to decrease the number of search options; we are prompting our users to draw the query structures precisely according to their needs (as much as possible) instead of modifying their requirements by extra search options using the same query structure.
Examples:
If uncharged targets are also expected as hits, the use of uncharged query is prompted instead of charged query structure and ignore charge search option.
| query | expected hit | search option in JOC | supported in JOC | supported in JPC |
|---|---|---|---|---|
| ignore charge | Yes | No | ||
| Yes | Yes | |||
| Yes | Yes |
If a single bond is required to match with an aromatic bond, the use of single or aromatic query bond is prompted, instead of the use of the vague bond level search option.
| query | expected hit | search option in JOC | supported in JOC | supported in JPC |
|---|---|---|---|---|
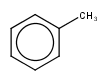 |
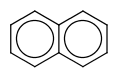 |
default vague bond level = 1 (in versions prior to 15.9.14) | Yes | No only vague bond level = 0 is available (in versions prior to 1.6) |
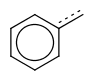 |
|
Yes | Yes |
Double bond stereo
JOC : With the exception of duplicate search, the default matching mode is ‘marked’; that is, only the stereo configuration of marked double bonds of the query structure are required to match with the double bonds of the target. In the case of non-marked double bonds ‘E’ matches ‘Z’, the doubleBondStereo parameter can be used to modify the behavior.
JPC : By default, ‘E’ does not match ‘Z’. We provide a transformation function, query_transform('query_structure', 'dbsmarkedonly'), which makes the double bond stereo search run similarly to JOC’s default.
Examples:
| Query | JOC default | JOCdoubleBondStereo:A | JPC default | JPCdbsmarkedonly |
|---|---|---|---|---|
| E or Z | E | E | E or Z |
(see doc: E/Z stereochemistry of double bonds)
Ligand pairs of a stereo double bond define a stereo configuration. (Referred to as cis/trans or E/Z configuration.) In 2D and 3D molecules this configuration is derived from the atomic coordinates.
We denote stereo configuration as:
-
Z: when the two atoms are on the same side of the double bond
-
E: when the two atoms are on the opposite sides of the double bond
Default interpretation of stereo notations in JPC
| Drawing | Interpretation |
|---|---|
| E | |
| Z | |
| E or Z | |
| E or Z | |
| Z | |
| E |
Using the query_transform(, 'dbsmarkedonly') function in JPC, you can change the default interpretation to:
| Drawing | Interpretation |
|---|---|
| E or Z | |
| E or Z | |
| E or Z | |
| E or Z | |
| Z | |
| E |
Ignore Tetrahedral Stereo information
J OC : there are more search options available for differently handling tetrahedral stereo information in searches.
JPC : By default, tetrahedral stereo information must be matched in substructure search. We provide a transformation function, query_transform('query_structure', 'ignoretetrahedralstereo') which makes possible to search without requiring tetrahedral stereo match. (available from version 5.1)
Higher Order Stereo information
JOC: Higher order stereo information can be taken into account in all search types (by default, they are ignored).
JPC : Higher order stereo is only supported in duplicate search, specifically these types of stereochemistry are affected: axial stereo, syn-anti stereo, and cumulene or ring cis-trans stereo. (from version 5.1)
Aromatization
(see doc: Aromatic conversion methods)
JOC : uses General aromatization method by default, but could be changed by applying the appropriate standardization method during indexing.
JPC : JPC uses molecule types stored in /etc/chemaxon/types/ folder. The column type of the chemical structures must be one of the molecule types present in this folder. The molecule type files can be created according to the needs of the user. The required standardizer actions - including the required aromatization method - can be defined there. The 'sample' molecule type - included in the installer - has General aromatization method.
SMILES/SMARTS Interpretation
In query structures, the default interpretation mode of molecule strings which can be ambiguously interpreted as SMILES and SMARTS is different.
| Query | JOC default SMARTS | JPC default SMILES |
|---|---|---|
| CCC |
Vague bond level / Bond matching
(see doc: Vague bond level)
JOC : supported Vague bond levels: n, h, 1, 2, 3, 4
default value = 1 (in versions prior to 15.9.14) Beyond aromatization three advanced features are also considered: handling of 5-membered rings with ambiguous aromaticity, 1-atom-long aromatic ring ligands and bridging bonds between two aromatic rings.)
default value = half ( from version 15.9.14) Beyond aromatization, the 5-membered rings are handled with ambiguous aromaticity.
JPC: worked on vague bond level: n (in versions prior to 1.6); that is bond types within the query structure are interpreted exactly as they are drawn; no other vague bond matching is available.
JPC works on vague bond level: half (from version 1.6). Beyond aromatization, the 5-membered rings are handled with ambiguous aromaticity.
Bond matching handling in JOC
JPC has no option to change bond match handling.
In JOC it is possible to choose between several levels of strictness in matching bond types, especially regarding aromaticity. The higher the level is, the more tolerant the bond matching becomes. For more details please visit our Vague bond level documentation.
Handling of 5-membered rings with ambiguous aromaticity
1-atom-long aromatic ring ligands
Bridging bonds between two aromatic rings
How to synchronize JOC behavior to JPC prior to version JOC 15.9.14 and JPC 1.6
It is not possible to change vague bond level in JPC. In JOC it is possible to change vague bond matching to the level of JPC by providing an option for the jc_compare function as shown in the example:
SELECT count(*) FROM nci_150k WHERE jc_compare(structure, 'Nc1ccccc1', 't:s vagueBond:n') = 1;Error handling
No result returned/structure not returned in JPC if format of structure is invalid/unknown. At other kind of errors you may create your own method to handle it according to your needs. A simple example to simulate never halt on any errors:
CREATE OR REPLACE FUNCTION chemterm_no_error(term text, mol Molecule) returns TEXT as
$$
BEGIN
RETURN chemterm(term,mol);
EXCEPTION
WHEN OTHERS THEN
RETURN NULL;
END;
$$
LANGUAGE plpgsql;Many methods of JOC have haltOnError option to determine what happens in case of errors.
Migration demo
See the video demonstrating migration from JChem Oracle Cartridge to JChem PostgreSQL Cartridge.